La validité biologique de nombreuses cibles de microARN remise en question
L'équipe d'Hervé Seitz à l’Institut de génétique humaine, met en évidence les sources des nombreux faux positifs produits par les méthodes expérimentales et informatiques couramment utilisées pour identifier les cibles de microARN. La plupart des cibles connues seraient en réalité trop peu réprimées par les microARN pour que cette régulation ait de réelles conséquences phénotypiques. Cette étude qui remet en cause les conclusions de nombreux travaux, a été publiée le 15 novembre 2016 dans la revue Genome Research.
Depuis la découverte de leur diversité il y a une quinzaine d’années, les microARN ont attiré une grande attention de la part de la communauté scientifique. Ces petits ARN régulent l’expression de gènes spécifiques, reconnus par complémentarité de séquence, le microARN s’appariant à un ARN messager cible dont il guide la répression. La reconnaissance de la cible est très tolérante vis-à-vis des imperfections de complémentarité entre les deux ARN, si bien que chaque microARN peut généralement se fixer à plusieurs centaines d’ARN messagers différents. C’est ainsi que les microARN ont acquis la réputation d’être de véritables « chefs d’orchestre » du génome, contrôlant directement plus de la moitié des gènes codants chez les Mammifères. On les soupçonne donc d’être impliqués dans tous les processus biologiques, en conditions saines ou pathologiques.
Deux outils principaux sont utilisés pour identifier les cibles de microARN :
- la biologie moléculaire, avec par exemple des expériences d’immuno-précipitation suivies de séquençage à haut débit pour identifier les ARN messagers interagissant avec les microARN dans un échantillon biologique donné;
- la bio-informatique pour rechercher les sites de complémentarité avec les microARN qui ont été conservés au cours de l’évolution.
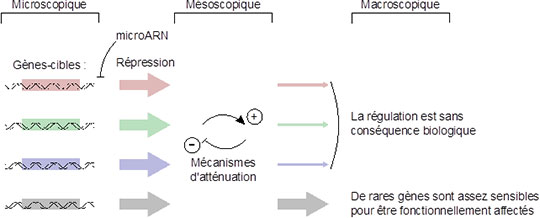
Les méthodes de biologie moléculaire ont l’avantage de ne pas faire appel à de nombreux présupposés théoriques, mais elles ont l’inconvénient de détecter toutes sortes d’interactions entre microARN et ARN messagers, y compris celles qui seraient sans conséquence macroscopique. En effet, les microARN répriment très peu leurs cibles (moins d’un facteur 2 en général). Or l’activité biologique de la plupart des gènes est robuste vis-à-vis d’aussi faibles fluctuations d’expression. Les mécanismes d’homéostasie, qui tendent à atténuer les variations de nombreux paramètres biologiques, annulent les conséquences macroscopiques des événements moléculaires d’intensité trop faible.
La bio-informatique est justement censée résoudre simplement ce problème : on considère qu’un élément génomique donné sera conservé au cours de l’évolution seulement s’il joue un rôle bénéfique à l’échelle de l’organisme. Dans le cas contraire, il devrait accumuler des mutations au même rythme que les éléments non-fonctionnels du génome. Grâce à la multiplication des séquençages de génomes entiers, il est devenu facile d’identifier les éléments génomiques phylogénétiquement conservés. On dispose maintenant d’alignements des séquences de génomes de nombreuses espèces, qui facilitent la recherche des sites de fixation de microARN conservés au cours de l’évolution.
Les chercheurs utilisent donc généralement une combinaison de ces deux types d’outils pour identifier des interactions observables expérimentalement, tout en s’assurant qu’elles jouent bien un rôle biologique en raison de leur conservation au cours de l’évolution.
Dans cette étude, l'équipe d'Hervé Seitz a identifié des sources de faux positifs inattendues dans les méthodes bio-informatiques.
D’une part, l'étude d’un microARN spécifique des neutrophiles (des cellules immunitaires), a confirmé pour la plupart des cibles prédites informatiquement un effet répresseur du microARN plus faible que la variation d’expression de ce gène entre individus sains de souche sauvage, et qui ne présentent pas de phénotype particulier. Ces fluctuations inter-individus ne sont donc pas suffisantes pour provoquer un phénotype macroscopique. Or pour la plupart des cibles prédites pour ce microARN, l’effet du microARN est encore plus faible que ces fluctuations.
D’autre part, les chercheurs ont démontré l’existence de deux problèmes conceptuels, à l’origine de faux positifs dans les prédictions informatiques.
1. Certains ARN messagers sont tellement abondants qu’ils peuvent séquestrer une grande proportion de la population d'un microARN donné. Ils peuvent donc moduler sa disponibilité pour les autres cibles. En d’autres termes, ces ARN messagers se comporteraient en régulateurs du régulateur. Il s’agirait donc d’une nouvelle fonction potentielle pour les sites d’interaction avec les microARN, susceptible d’être conservée au cours de l’évolution. Il est donc probable que certains sites de fixation de microARN soient conservés en raison de cette fonction de modulation des microARN, et non en raison de la répression de l’ARN messager par le microARN.
2. Par ailleurs, certaines régions du génome sont conservées pour des raisons indépendantes des microARN. Elles peuvent être par exemple des sites de fixation de protéines, et cette interaction serait bénéfique, donc conservée. Par hasard, ces régions peuvent être complémentaires de la séquence de microARN, ce qui n’est pas complètement inattendu, étant donnée la grande diversité des microARN répertoriés. Les programmes de prédiction de cibles de microARN, en repérant de tels sites conservés complémentaires des microARN, attribueraient la conservation phylogénétique à une hypothétique fonction de l’interaction entre l’ARN messager et le microARN – à tort, puisque la conservation serait due à un autre facteur. Les auteurs ont pu estimer la fréquence de ce type d’erreurs en dénombrant les sites de fixation aux microARN qui sont plus conservés que le microARN lui-même (par exemple, un microARN spécifique des Mammifères placentaires ne peut pas être responsable de la conservation d’un site de fixation conservé entre Mammifères et Poissons).
Cette étude remet donc en cause une bonne part des travaux précédents, qui tendent à considérer qu’une interaction entre microARN et ARN messager joue un rôle biologique du moment qu’elle est détectée et prédite informatiquement. Parmi les centaines de cibles qui ont été proposées pour chaque microARN, les auteurs proposent donc que seules quelques-unes (les gènes dont l’activité est la plus sensible vis-à-vis des petites fluctuations d’expression) sont véritablement des cibles, au sens biologique – les autres ne seraient que des événements moléculaires sans conséquence macroscopique.
© Hervé Seitz
En savoir plus
-
The number of biologically relevant microRNA targets has been largely overestimated
Natalia Pinzón, Blaise Li, Laura Martinez, Anna Sergeeva, Jessy Presumey, Florence Apparailly, and Hervé Seitz
Genome Res. Published in Advance November 15, 2016,doi:10.1101/gr.205146.116