Paysage génétique des tumeurs du sein: une histoire d’(im)maturité de la cellule d’origine
Les cancers du sein constituent une famille hétérogène de tumeurs, présentant des caractéristiques phénotypiques et génétiques différentes. Cette hétérogénéité est responsable de la résistance de certaines tumeurs aux traitements anticancéreux. L’origine de cette diversité reste cependant mal connue. Une équipe du Centre de recherche en cancérologie de Lyon, en étroite collaboration avec une équipe du Centre de recherche en cancérologie de Marseille, montre que l’état de différenciation de la cellule d’origine de la tumeur influence la réponse précoce à une activation oncogénique aberrante et détermine l’histoire génétique du processus de tumorigenèse. Cette étude a été publiée le 15 avril 2017 dans la revue Nature Medicine.
Le processus de tumorigenèse repose sur l'acquisition progressive d’anomalies génétiques et épigénétiques conférant un avantage sélectif de prolifération et de survie cellulaire. L’accumulation d’anomalies génétiques est rendue possible par la perte du maintien de l’intégrité du génome des cellules au cours du développement tumoral. A l’exception des tumeurs se développant sur la base d’un déficit de réparation de l’ADN, les mécanismes conduisant à la perte de stabilité du génome au cours de la transformation maligne et les éléments déterminant l’extension de l’instabilité dans une tumeur donnée restent à identifier.
De nombreuses observations indiquent que la survenue de l’instabilité génétique est un événement précoce au cours de la tumorigenèse et une conséquence directe des activations oncogéniques. En effet, la prolifération cellulaire induite par une activation mitogénique anormale provoque des lésions de l’ADN, incluant des cassures double-brin. La formation de ces cassures conduit à l’activation des voies dépendantes des kinases ATM et ATR et de la protéine onco-suppressive p53, à l’origine d’une sénescence prématurée ou d’une apoptose, deux puissantes barrières onco-suppressives. Cependant, la formation continue de dommages de l’ADN génère une forte pression de sélection sur l’inactivation de p53 et promeut l’acquisition d’une instabilité génétique, ouvrant la voie à la progression tumorale. Selon ce modèle, la survenue d’une activation mitogénique aberrante est donc à l’origine de deux caractéristiques fréquentes des cellules malignes : les mutations inactivatrices de p53 et l’instabilité génétique.
Les auteurs de cette étude ont cependant observé que certains cancers du sein triple négatifs (tumeurs agressives caractérisées par l’absence d’expression du récepteur aux œstrogènes, du récepteur à la progestérone et de l’oncoprotéine Her2/ErbB2), présentaient un nombre peu important d'altérations génomiques et une fréquence faible de mutations de p53, suggérant que le développement de ces tumeurs se produisait en absence d’instabilité génétique. Ils ont émis l’hypothèse selon laquelle ces caractéristiques seraient la conséquence des propriétés intrinsèques des cellules d’origine de ces tumeurs.
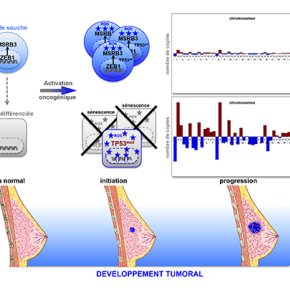
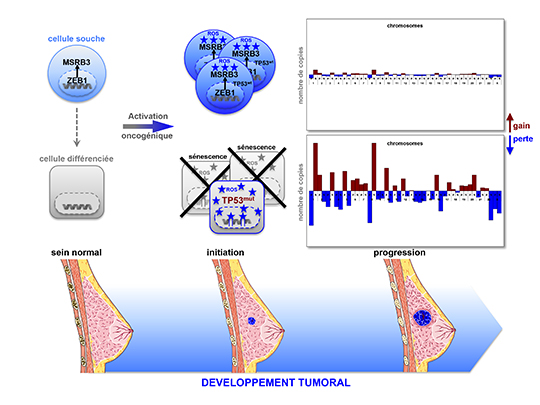
Afin de tester cette hypothèse, une hiérarchie des cellules épithéliales mammaires humaines normales a été établie et leur potentiel de différenciation a été évalué. La réponse des différentes sous-populations identifiées a ensuite été étudiée suite à l’induction d’une activation oncogénique par surexpression d’une forme mutée de RAS ou d’une surexpression de CYCLIN E, deux oncoprotéines à forte activité mitogénique. Alors que dans les cellules différenciées, cette activation induisait une formation précoce et massive de cassures de l’ADN, les sous-populations de cellules souches présentaient la capacité de soutenir l’activité mitogénique aberrante en absence de lésions de l’ADN. L'analyse de l'expression génique des sous-populations de cellules normales et de tumeurs du sein a permis de montrer que les cellules souches mammaires et les tumeurs avec une faible instabilité génomique présentaient pour caractéristique commune une expression élevée du facteur de transcription ZEB1. ZEB1 est un régulateur clef de la transition épithélio-mésenchymateuse, un processus de transdifférenciation embryonnaire, et de la plasticité cellulaire.
Les chercheurs ont ensuite montré que l’expression de ZEB1 dans les cellules épithéliales mammaires était suffisante pour induire des propriétés de cellules souches et pour prévenir la formation de lésions de l’ADN en réponse à une activation oncogénique. Son effet protecteur repose sur l’activation d’un système anti-oxydant conduit par la méthionine sulfoxide réductase MSRB3, cible transcriptionnelle de ZEB1. En prévenant la formation de cassures de l’ADN, l’expression de ZEB1 et de MSRB3 empêche l’activation de la voie dépendante de p53 et donc l’induction d’une apoptose ou d’une sénescence prématurée, facilitant ainsi la transformation maligne sur la base d’une faible instabilité génomique.
Au total, ces résultats montrent que l’expression de ZEB1 influence le comportement cellulaire en réponse à une activation oncogénique et que l'état de différenciation cellulaire détermine l’histoire génétique de la tumorigenèse mammaire.
© Roxane Pommier
En savoir plus
-
A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome stability.
Morel AP, Ginestier C, Pommier RM, Cabaud O, Ruiz E, Wicinski J, Devouassoux-Shisheboran M, Combaret V, Finetti P, Chassot C, Pinatel C, Fauvet F, Saintigny P, Thomas E, Moyret-Lalle C, Lachuer J, Despras E, Jauffret JL, Bertucci F, Guitton J, Wierinckx A, Wang Q, Radosevic-Robin N, Penault-Llorca F, Cox DG, Hollande F, Ansieau S, Caramel J, Birnbaum D, Vigneron AM, Tissier A, Charafe-Jauffret E, Puisieux A.
Nat Med. 2017 Apr 10. doi: 10.1038/nm.4323.