Le récepteur de la Ryanodine: une nouvelle cible thérapeutique contre la maladie d’Alzheimer
Des chercheurs de l’Institut de pharmacologie moléculaire et cellulaire, du laboratoire Physiologie et médecine expérimentale du cœur et des muscles et de l’université Columbia (USA), révèlent que des modifications post-traductionnelles du récepteur de la Ryanodine (RyR) participent au développement de la maladie d’Alzheimer. Ces travaux qui identifient la cascade moléculaire aboutissant à la dérégulation du RyR et démontrent qu’une prévention pharmacologique ou génétique de cette dérégulation réduit les symptômes de la maladie, ont été publiés le 16 juin 2017 dans la revue Journal of Biological Chemistry et le 19 juin 2017 dans la revue Acta Neuropathologica.
La maladie d’Alzheimer (MA) est une pathologie neurodégénérative qui représente un problème socio-économique et de santé publique majeur. Dans ce contexte, la compréhension des mécanismes à l’origine de la MA peut aider à identifier de nouvelles cibles cellulaires et développer de nouvelles stratégies thérapeutiques.
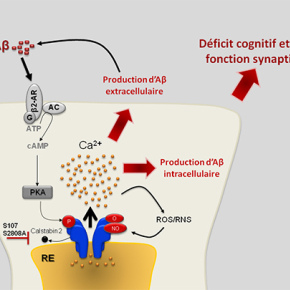
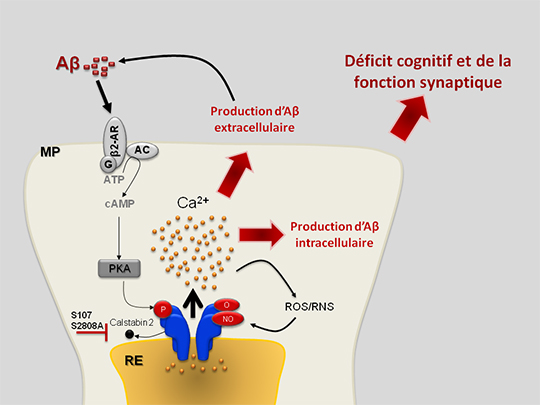
L’altération de l’homéostasie calcique du réticulum endoplasmique (RE), organelle de stockage du calcium intracellulaire, semble jouer un rôle central dans la MA. Dans deux publications complémentaires, les chercheurs démontrent l'implication du récepteur de la ryanodine (RyR), un canal de sortie de calcium du RE, dans le développement et l'amplification de la pathologie Alzheimer. Ils mettent en évidence pour la première fois que le RyR présente des modifications post-traductionnelles (MPTs) (hyperphosphorylation, oxydation, nitrosylation) dans des cerveaux humains issus de malades Alzheimer, dans deux modèles de souris transgéniques modèles de la MA (Lacampagne et al.), et dans un modèle cellulaire de la MA (Bussiere et al.). Ces MPTs du RyR entrainent la dissociation de la protéine régulatrice calstabine (FKBP12.6) du complexe macromoléculaire du RyR et l’augmentation de la fuite passive du calcium par le RyR et du stress oxydatif mitochondrial. L’utilisation d’un modèle d’étude in vitro leur a permis d’identifier la cascade moléculaire aboutissant à la dérégulation du RyR (Bussiere et al.). Ainsi, le peptide toxique amyloïde béta (Aβ) active la cacade β2-adrénergique induisant la phosphorylation du RyR. Les MPTs du RyR entrainent à leur tour une augmentation de la production de l’Aβ, constituant ainsi une boucle d’amplification de la MA.
Les résultats de ces travaux ont un impact clinique direct. En effet, la modulation pharmacologique par le composé S107 (qui stabilise la calstabine sur le complexe macromoléculaire du RyR), et génétique (croisement de souris transgéniques « Alzheimérisées » par l’expression de gènes portant des mutations familiales de la MA avec des souris transgéniques dont le gène du RYR est muté sur le site phosphorylé par la kinase PKA) permet de réduire la production de l’Aβ in vitro et in vivo et les déficits cognitifs et de plasticité synaptique chez les souris modèles de la MA.
Ces travaux révèlent que le RyR est un déterminant moléculaire majeur dans le développement et la progression de la MA. La stabilisation du complexe macromoléculaire du RyR pourrait constituer une nouvelle stratégie thérapeutique pour la maladie.
© Mounia Chami
En savoir plus
- Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease-like pathologies and cognitive deficits.
Lacampagne A, Liu X, Reiken S, Bussiere R, Meli AC, Lauritzen I, Teich AF, Zalk R, Saint N, Arancio O, Bauer C, Duprat F, Briggs CA, Chakroborty S, Stutzmann GE, Shelanski ML, Checler F, Chami M, Marks AR.
Acta Neuropathol. 2017 Jun 19. doi: 10.1007/s00401-017-1733-7
- Amyloid β production is regulated by β2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor.
Bussiere R, Lacampagne A, Reiken S, Liu X, Scheuerman V, Zalk R, Martin C, Checler F, Marks AR, Chami M.
J Biol Chem. 2017 Jun 16;292(24):10153-10168. doi: 10.1074/jbc.M116.743070. Epub 2017 May 5